138-2654-2846
138-2654-2846
美国EUA(紧急使用授权)认证的要点、用途及资料准备流程的全面解析,适用于医疗产品在公共卫生紧急状态下的快速上市
日期:2025-06-05 14:27
适用场景
仅在公共卫生紧急事件(如COVID-19、埃博拉)期间启动,由FDA根据《联邦食品、药品和化妆品法案》第564条授权。
常规替代:非紧急状态下需走FDA 510(k)、PMA或De Novo途径。
适用产品范围
| 类别 | 常见产品 | 有效期 |
|---|---|---|
| 体外诊断试剂 | 新冠病毒检测试剂盒(核酸/抗原) | 紧急状态结束+180天 |
| 治疗药物 | 瑞德西韦、单克隆抗体 | 紧急状态终止即失效 |
| 医疗器械 | 呼吸机、防护口罩(N95) | |
| 疫苗 | COVID-19疫苗(辉瑞/Moderna) |
授权标准
必要性:无其他已批准替代产品可用。
证据强度:现有数据证明产品收益大于风险(通常基于II期临床试验数据)。
合规基础:满足GMP/QSR质量体系要求。
加速产品上市
缩短审批时间至4-8周(常规FDA流程需6-18个月)。
案例:2020年首个COVID-19检测试剂EUA仅3天获批。
应对公共卫生危机
快速填补防疫物资缺口(如2020年N95口罩EUA授权中国制造商)。
临时市场独占性
在紧急状态期内,EUA产品可合法在美国销售/使用(医院、实验室等)。
为正式批准铺路
90%的COVID-19疫苗通过EUA上市后,1年内转为正式BLA批准。
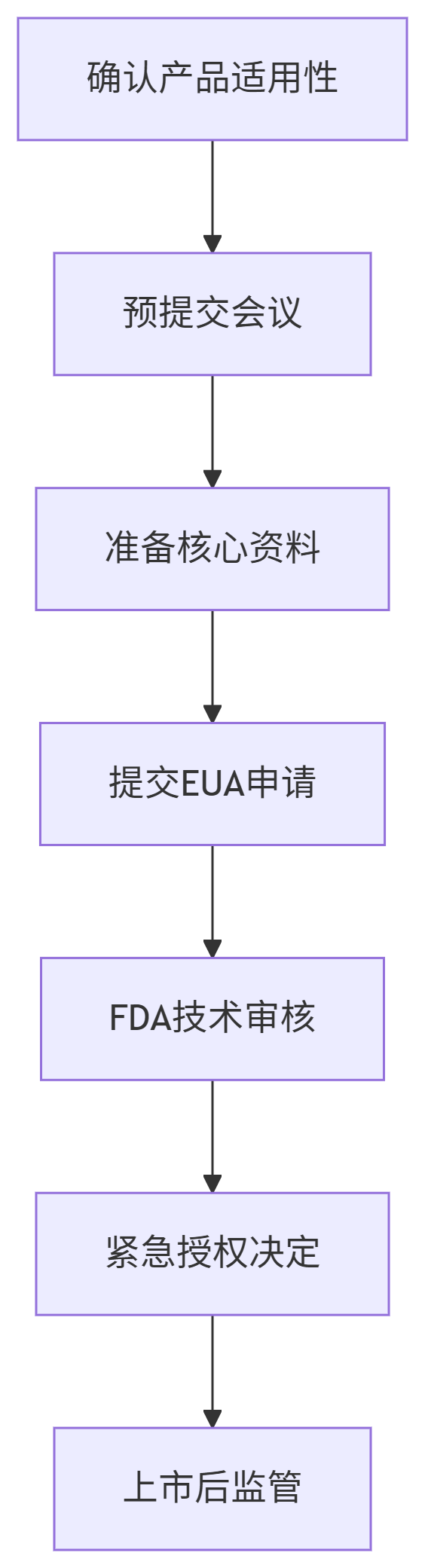
预提交阶段(Pre-submission)
向FDA提交Pre-EUA咨询包:产品说明书、性能验证方案、初步测试数据。
正式申请阶段(关键文件)
| 资料类型 | 具体要求 |
|---|---|
| 技术性能数据 | <ul><li>检测试剂:分析特异性/敏感性、LoD(检测限)数据</li><li>医疗器械:生物相容性、电气安全报告(IEC 60601)</li></ul> |
| 临床验证数据 | <ul><li>检测试剂:≥30阳性+30阴性样本的临床对比</li><li>治疗药物:II期临床试验报告</li></ul> |
| 质量体系文件 | <ul><li>ISO 13485证书</li><li>GMP合规声明(21 CFR Part 820)</li></ul> |
| 标签与说明书 | 必须标注:“仅限紧急使用,未获正式批准”(FDA模板) |
| 风险评估报告 | 包含假阳性/假阴性对公共卫生的影响分析 |
FDA审核阶段
补充临床样本数据(尤其变异毒株)
修改产品标签警示语
平均审核周期:15-30天(COVID-19检测试剂中位数为14天)。
常见补件要求:
上市后义务
每月提交不良事件报告(FAERS/MAUDE系统)。
持续监测产品性能(如检测试剂需跟踪变异株影响)。
| 维度 | EUA | 正式批准(510k/PMA) |
|---|---|---|
| 法律效力 | 临时授权(紧急状态有效) | 永久性上市许可 |
| 数据要求 | 有限临床证据(收益>风险) | 充分安全有效性数据(III期临床) |
| 申请成本 | $5万-$20万美元 | $50万-$300万美元 |
| 审批时间 | 1-2个月 | 6-36个月 |
授权撤销风险
若出现严重安全性问题(如检测试剂假阴性率>10%),FDA可立即撤销EUA。
案例:2021年撤销46个抗体检测试剂EUA。
紧急状态终止影响
EUA自动失效后,产品需在180天内退市,否则面临FDA罚没(最高$1M/单)。
责任豁免限制
不适用《PREP法案》全面免责,企业仍需承担产品缺陷责任。
📌 操作建议:通过FDA的 "Emergency Use Authorization (EUA) Submissions" 在线门户提交(需获取GUDID账户)。
预提交阶段(Pre-submission)
向FDA提交Pre-EUA咨询包:产品说明书、性能验证方案、初步测试数据。
正式申请阶段(关键文件)
| 资料类型 | 具体要求 |
|---|---|
| 技术性能数据 | <ul><li>检测试剂:分析特异性/敏感性、LoD(检测限)数据</li><li>医疗器械:生物相容性、电气安全报告(IEC 60601)</li></ul> |
| 临床验证数据 | <ul><li>检测试剂:≥30阳性+30阴性样本的临床对比</li><li>治疗药物:II期临床试验报告</li></ul> |
| 质量体系文件 | <ul><li>ISO 13485证书</li><li>GMP合规声明(21 CFR Part 820)</li></ul> |
| 标签与说明书 | 必须标注:“仅限紧急使用,未获正式批准”(FDA模板) |
| 风险评估报告 | 包含假阳性/假阴性对公共卫生的影响分析 |
FDA审核阶段
补充临床样本数据(尤其变异毒株)
修改产品标签警示语
平均审核周期:15-30天(COVID-19检测试剂中位数为14天)。
常见补件要求:
上市后义务
每月提交不良事件报告(FAERS/MAUDE系统)。
持续监测产品性能(如检测试剂需跟踪变异株影响)。
| 维度 | EUA | 正式批准(510k/PMA) |
|---|---|---|
| 法律效力 | 临时授权(紧急状态有效) | 永久性上市许可 |
| 数据要求 | 有限临床证据(收益>风险) | 充分安全有效性数据(III期临床) |
| 申请成本 | $5万-$20万美元 | $50万-$300万美元 |
| 审批时间 | 1-2个月 | 6-36个月 |
授权撤销风险
若出现严重安全性问题(如检测试剂假阴性率>10%),FDA可立即撤销EUA。
案例:2021年撤销46个抗体检测试剂EUA。
紧急状态终止影响
EUA自动失效后,产品需在180天内退市,否则面临FDA罚没(最高$1M/单)。
责任豁免限制
不适用《PREP法案》全面免责,企业仍需承担产品缺陷责任。
📌 操作建议:通过FDA的 "Emergency Use Authorization (EUA) Submissions" 在线门户提交(需获取GUDID账户)。
如果需要特定产品(如IVD试剂或呼吸机)的EUA申请模板,或最新FDA审核标准清单,可进一步提供详细附件。
如果你正在筹备出口业务,欢迎与东莞市安华检测技术有限公司直接沟通(0769-86057700/赵先生13826542846/13790607805)。
东莞市安华检测技术有限公司拥有自建实验室,为广大客户提供各行各业的检测认证服务,深受广大客户信赖。